Input files
APBS input files are loosely-formatted files which contain information about the input, parameters, and output for each calculation. These files are whitespace- or linefeed-delimited. Comments can be added to the input files via the # character; all text between the # and the end of the line is not parsed by APBS. Specific examples of APBS input are described in the Examples section.
APBS input files contain three basic sections which can be repeated any number of times:
READ: section for specifying input.
ELEC: section for specifying calculation parameters.
PRINT: section for specifying summary output.
These sections can occur in any order, however, they are clearly interdependent. For example, PRINT requires ELEC and ELEC requires one or more READ sections. Sections can also be repeated; several READ statements may be used to load molecules and multiple ELEC sections would specify various electrostatics calculations on one or more molecules.
READ statements
READ [keywords...]END
One of these sections must be present for every molecule involved in the APBS calculation. Molecule and "map" IDs are assigned implicitly assigned for each molecule/map read, based on order and starting at 1. This section has the following keywords:
mol {format} {path}
This command specifies the molecular data to be read into APBS. The arguments are:
- format
The format of the input data. Acceptable flags include:
- pqr
Molecular data is in PQR format (see Formats section)
- pdb
Molecular data is in PDB format.

We do not completely follow the PDB format, as specified in the above link. Specifically, we allow general whitespace-, tab-, or newline-delimited format, thereby permitting the manipulation of molecules with coordinates outside the �999 range. However, as a consequence of this flexibility, we cannot handle chain IDs in the PDB file!
- path
The location of the molecular data file.
parm {format} {path}
This command specifies the charge and radius data to be used with PDB-format molecule files. The arguments are:
- format
The format of the parameter file. Acceptable flags include:
- flat
APBS flat-file parameter format (see Formats section)
- path
The location of the parameter data file.
diel {format} {path-x} {path-y} {path-z}
This command allows APBS to read the dielectric function
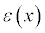 mapped to 3 meshes shifted by one-half grid spacing in the x, y, and z
directions. The result are maps of dielectric variables between the
solvent and biomolecular dieletric constants; these values are unitless.
In general, this command will read dielectric maps written by
write
commands in earlier APBS calculations.
Arguments for this command are:
mapped to 3 meshes shifted by one-half grid spacing in the x, y, and z
directions. The result are maps of dielectric variables between the
solvent and biomolecular dieletric constants; these values are unitless.
In general, this command will read dielectric maps written by
write
commands in earlier APBS calculations.
Arguments for this command are:
NOTE: if you choose this option and have a non-zero ionic strength, you must also include a read kappa statement
- format
The format of the dielectric map. Acceptable values include:
- dx
OpenDX format (see Formats section)
- path-x
The location of the x-shifted dielectric map file.
- path-y
The location of the y-shifted dielectric map file.
- path-z
The location of the z-shifted dielectric map file.
kappa {format} {path}
This command allows APBS to read the ion-accessibility function
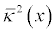 mapped to a mesh. The result are maps of ion accessibility values which
range between 0 and the build Debye-H�ckel screening parameter;
these values have units of �-2. In
general, this command will read kappa-maps written by
write
commands in earlier APBS calculations.
Arguments for this command are:
mapped to a mesh. The result are maps of ion accessibility values which
range between 0 and the build Debye-H�ckel screening parameter;
these values have units of �-2. In
general, this command will read kappa-maps written by
write
commands in earlier APBS calculations.
Arguments for this command are:NOTE: if you choose this option, you must also include a read diel statement
- format
The format of the kappa map. Acceptable values include:
- dx
OpenDX format (see Formats section)
- path
The location of the kappa map file.
charge {format} {path}
This command allows APBS to read the fixed (molecular) charge density function mapped to a mesh. The result are maps of charge densities; these values have units of ec (electron charge). In general, this command will read charge-maps written by write commands in earlier APBS calculations. Arguments for this command are:
- format
The format of the charge map. Acceptable values include:
- dx
OpenDX format (see Formats section)
- path
The location of the charge map file.
ELEC statements
ELEC [name id] {type} [keywords...]END
This section is the main component of all APBS runs. There may be several ELEC sections, operating on different molecules or using different parameters for multiple runs on the same molecule. The order of the ELEC statement matters (see above); the arguments are:
- name id
This optional command allows users to assign an alphanumeric string to the calculation to facilitate later operations (particularly in the PRINT statements).
- type
Specify the type of electrostatics calcultion to perform (these are described in greater detail below):
mg-auto for automatically-configured sequential focusing multigrid calculations.
mg-para for automatically-configured parallel focusing multigrid calculations.
mg-manual for manually-configured multigrid calculations.
fe-manual for manually-configured adaptive finite element calculations.
mg-dummy for calculations of surface and charge distribution properties which do not require solution of the PBE.
- keywords
Keywords describing the parameters of the electrostatic calcualtion. These are described in the Keywords section below.
Automatic sequential focusing multigrid calculation (mg-auto)
This automatically sets up and performs a string of single-point PBE calculations to "focus" on a region of interest (binding site, etc.) in a system. It is basically an automated version of mg-manual designed for easier use. Most users should probably use this version of ELEC.
The keywords for this command (described in more detail in the Keywords section) are listed below. All keywords are required (no default values!) unless otherwise noted: dime, cglen, fglen, cgcent, fgcent, mol, lpbe or npbe, bcfl, ion (optional), pdie, sdie, chgm, usemap (optional), sdens, srad, swin, temp, gamma, calcenergy, calcforce, write, writemat
Automatic parallel focusing multigrid calculation (mg-para)
This calculation closely resembles mg-auto in syntax. However, it is basically designed to perform single-point calculations on systems in a parallel focusing fashion. While this method does provide support for decreasing the domain size from a coarse (large) global grid to a fine (smaller) global grid, it should not be used to look at subsets of biomolecules such as titration sites, etc. Such subset calculations require more complicated energy evaluation which is not yet supported by mg-para. However, since parallel focusing was designed to provide detailed evaluation of the electrostatic potential on a large scale, such subset calculations are better left to traditional focusing via the mg-auto keyword.
The keywords for this command (described in more detail in the Keywords section) are listed below. All keywords are required (no default values!) unless otherwise noted: dime, ofrac, pdime, async, cglen, fglen, cgcent, fgcent, mol, lpbe or npbe, bcfl, ion (optional), pdie, sdie, chgm, usemap (optional), sdens, srad, swin, temp, gamma, calcenergy, calcforce, write, writemat
Manual multigrid calculation (mg-manual)
This is the standard single-point PBE calculation performed by most solvers. The mg-manual calculation offers the most control of parameters to the user. Several of these calculations can be strung together to perform focusing calculations by judicious choice of the bcfl flag, however, the setup of the focusing is not automated as it is in mg-auto and mg-para calculations and therefore this command should only be used by more experienced users.
The keywords for this command (described in more detail in the Keywords section) are listed below. All keywords are required (no default values!) unless otherwise noted: dime, nlev, glen or grid, gcent, mol, lpbe or npbe, bcfl, ion (optional), pdie, sdie, chgm, usemap (optional), sdens, srad, swin, temp, gamma, calcenergy, calcforce, write, writemat
Manual adaptive finite element calculation (fe-manual)
This is a single-point PBE calculation performed by our adaptive finite element PBE solver. It requires that APBS was linked to the Holst group FEtk finite element library (http://www.fetk.org) during compilation.
The finite element solver uses a "solve-estimate-refine" cycle. Specifically, starting from an initial mesh, it performs the following iteration:
solve the problem
estimate the error in the solution
adaptively refine the mesh
| These methods are most useful for a select set of problems which can benefit from adaptive refinement of the solution. Furthermore, this implementation is experimental. In general, the sequential and parallel focusing multigrid methods offer the most efficient solution of the PBE for most systems |
The keywords for this command (described in more detail in the Keywords section) are listed below. All keywords are required (no default values!) unless otherwise noted: glen, etol, ekey, akeyPRE, akeySOLVE, targetNum, targetRes, maxsolve, maxvert, mol, lpbe or npbe or lrpbe or nrpbe, bcfl, ion (optional), pdie, sdie, chgm, usemap (optional), sdens, srad, swin, temp, gamma, calcenergy, calcforce, write, writemat
Manual non-numerical calculations (mg-dummy)
This allows users to write out dielectric, ion-accessibility, and charge distribution maps based on biomolecular geometry without actually solving the PB equation. The syntax is identical to mg-dummy.
The keywords for this command (described in more detail in the Keywords section) are listed below. All keywords are required (no default values!) unless otherwise noted: dime, nlev, cglen or grid, gcent, mol, lpbe or npbe, bcfl, ion (optional), pdie, sdie, chgm, usemap (optional), sdens, srad, swin, temp, gamma, calcenergy, calcforce, write, writemat
Keyword descriptions
This is a list of keywords used in the ELEC statements of APBS. Note that not all keywords are used in every ELEC statement; see above.
akeyPRE {key}
Specify how the initial finite element mesh should be constructed (from refinement of a very coarse 8-tetrahedron mesh prior to the solve-estimate-refine iteration. This allows for various a priori refinement schemes.
- key
The method used to guide initial refinement:
- unif
Uniform refinement
- geom
Geometry-based refinement at molecular surfaces and charges
akeySOLVE {key}
Specify how the the finite element mesh should be adaptively subdivided during the solve-estimate-refine iterations. This allows for various a posteriori refinement schemes.
- key
The method used to guide adpative refinement:
- resi
Residual-based a posteriori refinement
async { rank }
This optional keyword allows users to perform the different tasks in a parallel run asynchronously. Specifically, a processor masquerades as process rank in a parallel focusing run and provides output (data files and energies/forces) appropriate to that processor's local partition. The user must then assemble the results after all processes complete. First, this option is useful for scheduling on-demand resources: this makes it easy for users to backfill into the available processes in a queue. Second, this option is useful for running on limited resources: this enables users without access to large parallel machines to still perform the same calculations.
- rank
The ID of the particular processor to masquerade as. Processor IDs range from 0 to N-1, where N is the total number of processors in the run (see pdime). Processor ranks are related to their position in the overall grid by
where nx is the number of processors in the x-direction, ny is the number of processors in the y-direction, nz is the number of processors in the z-direction, i is the index of the processor in the x-direction, j is the index of the processor in the y-direction, k is the index of the processor in the z-direction, and p is the overall rank of the processor.
bcfl {flag}
Specify the type of boundary conditions used to solve the Poisson-Boltzmann equation:
- flag
The flag specifying the boundary condition definition:
- zero
"Zero" boundary condition. Potential at boundary is set to zero. This condition is not commonly used and can result in large errors if used inapprpriately.
- sdh
"Single Debye-H�ckel" boudnary condition. Potential at boundary is set to the values prescribed by a Debye-H�ckel model for a single sphere with a point charge. The sphere radius is set to the radius of the biomolecule and the sphere charge is set to the total charge of the protein. This condition works best when the boundary is sufficiently far from the biomolecule.
- mdh
"Multiple Debye-H�ckel" boundary condition. Potential at boundary is set to the values prescribed by a Debye-H�ckel model for a multiple, non-interacting spheres with a point charges. The sphere radii are set to the atomic radii of the biomolecule and the sphere charges are set to the total charge of the protein. This condition works better than sdh for closer boundaries but can be very slow for large biomolecules.
- focus
"Focusing" boundary condition. Potential at boundary is set to the values computed by the previous (usually lower-resolution) PB calculation. This is used in sequential focusing performed manually in mg-manual calculations. All of the boundary points should lie within the domain of the previous calculation for best accuracy; if any boundary points lie outside, their values are computed using single Debye-H�ckel boundary conditions (see above).
calcenergy { flag }
This optional keyword controls electrostatic energy output from a PBE calculation.
Note that this option must be used consistently for all calculations that will appear in subsequent PRINT statements. For example, if the statement print energy 1 - 2 end appears in the input file, then both calculations 1 and 2 must have calcenergy keywords present with the same values for flag.
- flag
Specify the types of energy values to be returned:
- no
(Deprecated) don't calculate any energies.
- total
Calculate and return total electrostatic energy for the entire molecule.
- comps
Calculate and return total electrostatic energy for the entire molecule as well as electrostatic energy components for each atom.
calcforce { flag }
This optional keyword controls electrostatic and apolar force output from a PBE calculation.
Note that this option must be used consistently for all calculations that will appear in subsequent PRINT statements. For example, if the statement print force 1 - 2 end appears in the input file, then both calculations 1 and 2 must have calcforce keywords present with the same values for flag.
- flag
Specify the types of force values to be returned:
- no
(Deprecated) don't calculate any forces.
- total
Calculate and return total electrostatic and apolar forces for the entire molecule.
- comps
Calculate and return total electrostatic and apolar forces for the entire molecule as well as force components for each atom.
cgcent {mol id | xcent ycent zcent}
Specify the center of the coarse grid (in a focusing calculation) based on a molecule's center or absolute coordinates. The arguments for this keyword are:
- mol id
Center the grid on molecule with ID id; as assigned in the READ section. Molecule IDs are assigned in the order they're read, starting at 1.
- xcent ycent zcent
The coordinates (in �) at which the grid is centered. Based on the PDB coordinate frame.
cglen {xlen ylen zlen}
Specify the coarse mesh domain lengths in a focusing calculation; this may be different in each direction. This is the starting mesh, so it should be large enough to complete enclose the biomolecule and ensure that the chosen boundary condition (see bcfl) is appropriate.
- xlen ylen zlen
Grid lengths in the x-, y-, and z-directions in �.
chgm {flag}
Specify the method by which the biomolecular point charges (i.e., Dirac delta functions) are mapped onto the grid. As we are attempting to model delta functions, the support (domain) of these discretized charge distributions is always a function of the grid spacing.
- flag
Discretization method (options have multiple declarations for backward-compatibility):
- spl0
Traditional trilinear interpolation (linear splines). The charge is mapped onto the nearest-neighbor grid points. Resulting potentials are very sensitive to grid spacing, length, and position.
- spl2
Cubic B-spline discretization. The charge is mapped onto the nearest- and next-nearest-neighbor grid points. Resulting potentials are somewhat less sensitive (than spl0) to grid spacing, length, and position.
dime {nx ny nz}
Number of grid points for grid-based discretization. For mg-manual, the arguments are dependent on the choice of nlev by the formula
where n is the dime argument, c is a non-zero integer, l is the nlev value. The most common values for grid dimensions are 65, 97, 129, and 161 (they can be different in each direction); these are all compatible with a nlev value of 4. If you happen to pick a "bad" value for the dimensions (i.e., mismatch with nlev), the code will adjust the specified dime downwards to more appropriate values. This means that "bad" values will typically result in lower resolution/accuracy calculations! The arguments for this keyword are:- nx ny nz
Number of grid points in the x-, y-, and z-directions.
ekey { flag }
Specify the method used to determine the error tolerance in the solve-estimate-refine iterations of the finite element solver.
- flag
Tolerance is interpreted as...
- simp
...per-simplex error limit
- global
...global (whole domain) error limit
- frac
...fraction of simplices you'd like to see refined
etol { tol }
Specify the tolerance for error-based adaptive refinement during the solve-estimate-refine iterations of the finite element solver. See also: ekey.
- tol
The error tolerance for adaptive finite element refinement. The exact definition of this tolerance is determined by the value of ekey.
fgcent {mol id | xcent ycent zcent}
Specify the center of the fine grid (in a focusing calculation) based on a molecule's center or absolute coordinates. The arguments for this keyword are:
- mol id
Center the grid on molecule with ID id; as assigned in the READ section. Molecule IDs are assigned in the order they're read, starting at 1.
- xcent ycent zcent
The coordinates (in �) at which the grid is centered. Based on the PDB coordinate frame.
fglen {xlen ylen zlen}
Specify the fine mesh domain lengths in a focusing calculation; this may be different in each direction. This should enclose the region of interest in the molecule.
- xlen ylen zlen
Grid lengths in the x-, y-, and z-directions in �.
gamma { value }
The coefficient (surface tension) for solvent-accesisble surface area (SASA) models of apolar forces. This term only enters into force calculations; SASA-based energies must be computed separately (see acc).
- value
SASA-based apolar coefficient (in kJ/mol/�).
gcent {mol id | xcent ycent zcent}
Specify the center of the grid based on a molecule's center or absolute coordinates. The arguments for this keyword are:
- mol id
Center the grid on molecule with ID id; as assigned in the READ section. Molecule IDs are assigned in the order they're read, starting at 1.
- xcent ycent zcent
The coordinates (in �) at which the grid is centered. Based on the PDB coordinate frame.
glen {xlen ylen zlen}
Specify the mesh domain lengths; this may be different in each direction. For some invocations of APBS, either this key or grid must be specified.
- xlen ylen zlen
Grid lengths in the x-, y-, and z-directions in �.
grid {hx hy hz}
Specify the mesh grid spacings; this may be different in each direction. For some invocations of APBS, either this key or glen must be specified.
- hx hy hz
Grid spacings in the x-, y-, and z-directions in �.
ion {charge} {conc} {radius}
Specify the mobile ion species present in the system. This command can be repeated as necessary to specify multiple types of ions; however, only the largest ionic radius is used to determine the ion-accessibility function.
- charge
Mobile ion species charge (in ec)
- conc
Mobile ion species concentration (in M)
- radius
Mobile ion species radius (in �)
lpbe
Specifies that the linearized PBE should be solved. Either this keyword or npbe, nrpbe, or lrpbe, must be present (based on the calculation type).
lrpbe
Specifies the linearized form of the regularized PBE equation (RPBE). The regularized PBE equation replaces the point charge distribution with the corresponding Green's function. As a result of this replacement, the solution corresponds to the reaction field instead of the total potential; the total potential can be recovered by adding the approrriate Coulombic terms to the solution. Likewise, this equation immediately yields the solvation energy without the need for reference calculations. Either this keyword or lpbe, npbe, or nrpbe, must be present (based on the calculation type).
This function is only available for FEM-based solvers.
maxsolve { num }
Specify the number of times to perform the solve-estimate-refine iteration of the finite element solver. See also: maxvert, targetRes,
- num
Maximum number of iterations.
maxvert { num }
Specify the maximum number of vertices to allow during solve-estimate-refine cycle of finite element solver. This places a limit on the memory that can be used by the solver. See also: targetRes, maxsolve.
- num
Maximum number of vertices.
mol {id}
Specify the molecule for which the PBE is to be solved. IDs are based on the order in which molecules are read by read mol statements, starting from 1.
- id
The ID of the molecule for which the PBE is to be solved.
nlev {lev}
Specify the depth of the multilevel hierarchy used in the multigrid solver. See dime for a discussion of how nlev relates to grid dimensions.
- lev
Depth of the multigrid hierarchy.
npbe
Specifies that the nonlinear (full) PBE should be solved. Either this keyword or lpbe, nrpbe, or lrpbe, must be present (based on the calculation type).
nrpbe
Specifies the nonlinear form of the regularized PBE equation (RPBE). The regularized PBE equation replaces the point charge distribution with the corresponding Green's function. As a result of this replacement, the solution corresponds to the reaction field instead of the total potential; the total potential can be recovered by adding the approrriate Coulombic terms to the solution. Likewise, this equation immediately yields the solvation energy without the need for reference calculations. Either this keyword or lpbe, npbe, or nrpbe must be present (based on the calculation type).
This function is only available for FEM-based solvers.
pdie {diel}
Specify the dielectric constant of the biomolecule. This is usually a value between 2 to 20, where lower values consider only electronic polarization and higher values consider additional polarization due to intramolecular motion.
- diel
Biomolecular dielectric constant (unitless)
pdime {npx npy npz}
Specify the processor array to be used in a parallel focusing calculation. The product npx � npy � npz should be less than or equal to the total number of processors with which APBS was invoked (usually via mpirun). If more processors are provided at invocation than actually used during the run, the extra processors are not used in the calculation. The processors are tiled across the domain in a Cartesian fashion with a specified amount of overlap (see ofrac) between each processor to ensure continuity of the solution.
- npx npy npz
The number of processors to be used in the x-, y- and z-directions of the system.
ofrac {frac}
Specify the amount of overlap to include between the individual processors meshes in a parallel focusing calculation (see ofrac). This should be a value between 0 and 1.
- frac
Amount of overlap between processor meshes; a value between 0 and 1.

Empirical evidence suggests that an ofrac value of 0.1 is sufficient to generate stable energies. However, this value may not be sufficient to generate stable forces and/or good quality isocontours. For example, the following table illustrates the change in energies and visual artifacts in isocontours as a function of ofrac values for a small peptide (2PHK:B).
sdie {diel}
Specify the dielectric constant of the solvent. Bulk water at biologically-relevant temperatures is usually modeled with a dielectric constant of 78-80.
- diel
Solvent dielectric constant (unitless)
sdens {density}
Specify the number of grid points per square-angstrom to use in Vacc object. Ignored when srad is 0.0 (see srad) or srfm is spl2 (see srfm). There is a direct correlation between the value used for the Vacc sphere density, the accuracy of the Vacc object, and the APBS calculation time. APBS default value is 10.0.
- density
Vacc sphere density (in grid points/�2 ).
srad {radius}
Specify the radius of the solvent molecules; this parameter is used to define the dielectric function (see srfm). This value is usually set to 1.4 � for water.
- radius
Solvent radius (in �).
swin {win}
Specify the size of the support (i.e., the rate of change) for spline-based surface definitions (see srfm). Usually 0.3 �.
- win
Spline window (in �).
srfm {flag}
Specify the model used to construct the dielectric ion-accessibility coefficients.
- flag
The coefficient model:
- mol
The dielectric coefficient
is defined based on
a molecular surface definition. The problem domain is
divided into two spaces. The "free volume" space is
defined by the union of solvent-sized spheres (see
srad)
which do not overlap with biomolecular atoms. This
free volume is assigned bulk solvent dielectric values.
The complement of this space is assigned biomolecular
dielectric values. With a non-zero solvent radius
(srad), this choice of coefficient
corresponds to the traditional definition used for PB
calculations. When the solvent radius is set to zero,
this corresponds to a van der Waals surface
definition.The ion-accessibility coefficient
is defined by an "inflated" van der Waals model.
Specifically, the radius of each biomolecular atom is
increased by the radius of the ion species. The
problem domain is then divded into two spaces. The
space inside the union of these inflated atomic spheres
is assigned an ion-accessibility value of 0; the
complement space is assigned bulk ion accessibility
values.
- smol
The dielectric and ion-accessiblity coefficients are defined as for mol (see above). However, they are then "smoothed" by a 9-point harmonic averaging to somewhat reduce sensitivity to the grid setup as described by Bruccoleri et al. J Comput Chem 18 268-276, 1997 (doi: 10.1002/(SICI)1096-987X(19970130)18:2<268::AID-JCC11>3.0.CO;2-E).
- spl2
The dielectric
and ion-accessibility
coefficients are defined by a cubic-spline surface as
described by Im et al, Comp Phys
Commun 111 (1-3) 59-75, 1998
(doi:10.1016/S0010-4655(98)00016-2).
These spline-based surface definitions are very stable
with respect to grid parameters and therefore ideal for
calculating forces. However, they require substantial
reparameterization of the force field; interested users
should consult Nina et al, Biophys
Chem 78 (1-2) 89-96, 1999
(doi:10.1016/S0301-4622(98)00236-1).
Additionally, these surfaces can generate unphysical
results with non-zero ionic strengths; this is an
on-going area of development.
targetRes { res }
Specify the target resolution of the simplices in the mesh; refinement will continue until the longest edge of every simplex is below this value. See also: maxvert, maxsolve, targetNum
- res
Target resolution for longest edges of simplices in mesh (in �).
targetNum { num }
Specify the target number of vertices in the initial mesh; intiial refinement will continue until this number is reached or the the longest edge of every simplex is below targetNum. See also: targetRes
- num
Target number of vertices in initial mesh.
temp { T }
Temperature for PBE calculation.
- T
Temperature (in K)
usemap {type} {id}
Specify pre-calculated coefficient maps to be used in the PB calculation. These must have been input via an earlier read map statement.
- type
Specify the type of pre-calculated map to be read in:
- diel
Dielectric function map (as read by read map dielx diely dielx); this causes the pdie, sdie, srad, swin, and srfm parameters and the radii of the biomolecular atoms to be ignored when computing dielectric values for the PBE.
- kappa
Mobile ion-accessibility function map (as read by read map kappa); this causes the swin and srfm parameters and the radii of the biomolecular atoms to be ignored when computing mobile ion values for the PBE. The ion parameter is not ignored and will still be used.
- charge
Charge distribution map (as read by read map charge); this causes the chgm parameter and the charges of the biomolecular atoms to be ignored when assembling the fixed charge distribution for the PBE.
- id
This ID specifies the particular map read in with read map. These IDs are assigned sequentially, starting from 1, for each map type read by APBS.
write {type} {format} {stem}
This controls the output of scalar data calculated during the PB run. This keyword can be repeated several times to provide various types of data.
- type
What type of data to output:
- charge
Write out the biomolecular charge distribution in units of ec (multigrid only)
- pot
Write out the electrostatic potential in units of kbT/ec (multigrid and finite element)
- smol
Write out the solvent accessibility defined by the molecular surface definition (see srfm smol). Values are unitless and range from 0 (inaccessible) to 1 (accessible). (multigrid and finite element)
- sspl
Write out the spline-based solvent accessibility (see srfm spl2). Values are unitless and range from 0 (inaccessible) to 1 (accessible). (multigrid and finite element)
- vdw
Write out the van der Waals-based solvent accessibility (see srfm smol with srad smol). Values are unitless and range from 0 (inaccessible) to 1 (accessible). (multigrid and finite element)
- ivdw
Write out the inflated van der Waals-based ion accessibility (see srfm smol). Values are unitless and range from 0 (inaccessible) to 1 (accessible). (multigrid and finite element)
- lap
Write out the Laplacian of the potential
in units of kBT/ec/�2 (multigrid only).- edens
Write out the "energy density"
in units of kBT/ec/�2 (multigrid only).- ndens
Write out the mobile ion number density
for m ion species in units of M (multigrid only).- qdens
Write out the mobile charge density
for m ion species in units of ec M (multigrid only).- dielx
Write out the dielectric map shifted by 1/2 grid spacing in the x-direction (see read diel). The values are unitless (multigrid only).
- diely
Write out the dielectric map shifted by 1/2 grid spacing in the y-direction (see read diel). The values are unitless (multigrid only).
- dielz
Write out the dielectric map shifted by 1/2 grid spacing in the z-direction (see read diel). The values are unitless (multigrid only).
- kappa
Write out the ion-accessibility kappa map (see read kappa). The values are in units of �-2 (multigrid only).
- format
Specify the format for writing out the data.
- dx
Write out data in OpenDX format. This is the preferred format for APBS I/O. (multigrid and finite element).
- avs
Write out data in AVS UCD format. (finite element only)
- uhbd
Write out data in UHBD format. (multigrid only)
- stem
Specify the path for the output; files are written to stem.XXX, where XXX is determined by the file format (and processor rank for parallel calculations).
write {type} {stem}
This controls the output of the mathematical operators in the PBE as matrices in Harwell-Boeing format (multigrid only)
- type
What type of operator to output:
- poission
Write out the Poisson operator.
- pot
Write out the Gateaux (functional) derivative of the full PBE operator evaluated at the current solution.
- stem
Specify the path for the output; files are written to stem.mat.
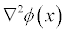
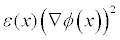
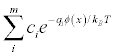
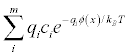
PRINT statements
PRINT {what} [id op id op...]END
This is a very simple section that allows linear combinations of calculated properties to be written to standard output. It has the following variables:
- what
Specify which quantities to manipulate/print:
- energy
Print energies as calculated with an earlier calcenergy ELEC command.
- force
Print forces as calculated with an earlier calcforce ELEC command.
- id
The ID of a particular ELEC calculation as specified with the ELEC name id command. If the id variables are not set explicitly, they are assigned sequential integers, starting at 1, based on the order of the ELEC statements.
- op
Specify the arthimetic operation to be performed on the calculated quantities:
- +
Addition
- -
Subtraction
Given all these options, a typical declaration might look like:
Example 2. PRINT statement example
# Energy change due to binding
print energy complex - ligand - protein end
# Energy change due to solvation
print energy solvated - reference end
# Solvation energy change due to binding
print energy
complex_solv - complex_ref
- ligand_solv + ligand_ref
- protein_solv + protein_ref
end
|
See the APBS examples/ directory for more examples.