
The basic function of X-Score is to compute the binding score of a given ligand molecule (or multiple ligand molecules) to a target protein. The protein is required to be stored in a PDB file, and the ligand molecule(s) should be stored in a Sybyl/Mol2 file. All the parameters needed to run X-Score are assembled in an input file (click here to view an example). You are supposed to edit this file to meet your own purpose.
To run X-Score, simply use this file as input:
Example: xscore name_of_the_input_file
The parameters specified in the input file will be explained in detail below. You can find an example input file under the "example/" directory.
2. How to prepare the protein in PDB format?
X-Score needs the three-dimensional structure of the protein-ligand complex to calculate its binding constant. The structure could be either experimentally determined or modeled by a docking program. Since most today's molecular docking programs will keep the protein rigid while docking the ligand(s), for the sake of efficient computing, X-Score requires the protein and the ligand(s) to be stored in two separate files.
The three-dimensional structure of the protein is supposed to be stored in the PDB format. When preparing this PDB file, please remember: (1) Add polar hydrogen atoms. Add all hydrogen atoms will not hurt but only polar hydrogen atoms are needed in computation. (2) Remove any ligand or other organic cofactors. (3) Remove all the water molecules. (4) If a metal ion exists inside the binding site and is believed to be important for ligand binding, keep it as part of the protein. X-Score considers this kind of metal ions in computation. According to the PDB convention, a metal ion should be described by a line started by "HETATM". Please be cautious because not every software writes standard PDB file! Some of them, such as SYBYL v6.8, are even unable to export metals to a PDB file. If this happens, you may want to manually add a line for the metal to the PDB file. (5) Occasionally, there are some non-natural residues on the protein, X-Score will neglect them if it cannot find appropriate parameters for them in the parameter libraries.
The parameter RECEPTOR_PDB_FILE in the input file specifies the path and the name of this file.
Sometimes there are an organic cofactor binding together with the ligand molecule inside the binding pocket, such as CoA, NADH, and etc., and maybe you want to keep it at its place when evaluating binding affinities of the ligand molecules. X-Score does provide such an option: it will treat the cofactor as part of the protein. Since this kind of cofactors are usually not formed by any standard building blocks, the PDB format is not proper for presenting them. But for instead, you can save the cofactor in SYBYL MOL2 format and specify its path and name with the COFACTOR_MOL2_FILE parameter in the input file. Note that: (1) Atom types and bond types in this molecule should be assigned correctly according to SYBYL's definition. (2) All hydrogen atoms should be added onto it. (3) This cofactor molecule should share the same coordinate system with the protein and the ligand. (4) If the cofactor is originally covalently bound to the protein, you need to remove that bond to keep the cofactor as a separate molecule. This modification will not affect X-Score's computation.
3. How to prepare the ligands in Mol2 format?
The primary thing that should be kept in mind is: the ligand molecules must be pre-docked into the binding pocket of the target protein. X-Score is not a docking program. It can only calculate the binding affinities for given protein-ligand complexes.
X-Score requires the ligands to be stored in the SYBYL Mol2 format. Naturally, we recommend SYBYL for such preparation. Please make sure that the atom type and the bond type assignments are correct according to the Tripos force field conventions. X-Score is able to identify and correct some common errors in atom typing and bond typing but certainly cannot handle all the possible situations. Other molecular modeling software may also support the Mol2 format. There are also some programs, such as Babel, which are specially designed for converting different formats. However, our experience is that format conversion is not always carried out in a flawless way.
All hydrogen atoms need to be added to the ligand molecules. Atomic charges are not necessary for X-Score computation.
If there is more than one ligand molecule, all of them should be packed one after one in this file. This is often referred as a "multiple" Mol2 file. Since handling a very large file will probably slow down your computer significantly, we do not recommend you to pack too many molecules in one file. A generally acceptable limit is 100,000 molecules (approximately several hundred MB in terms of the file size). If you have to process even more ligands, you may split them into several Mol2 files and run X-Score for each of them respectively.
The parameter LIGAND_MOL2_FILE in the input file specifies the path and the name of this file.
4. How to set the other parameters in the input file?
In the input file, all the lines started with "#" are notations and are neglected by the program.
The parameter FUNCTION should be set to "SCORE". This tells the program to perform X-Score computation.
As we have mentioned in the Introduction section, there are three scoring functions implemented in X-Score, i.e. HPScore, HMScore, and HSScore. You can find three switches in the input file, i.e. APPLY_HPSCORE, APPLY_HMSCORE, and APPLY_HSSCORE. You may set any of them as "YES" or "NO" to choose the combination you like. If all the three scoring functions are switched on, typically X-Score can process ~10,000 molecules an hour on an SGI Octane2/R12000/360MHz workstation.
Another feature of X-Score is the option of pre-screening the ligands by molecular properties. This is also well known as "Lipinski rules" in drug design, which are some crude judgments of "drug-likeness". Many approaches have suggested that, by applying such chemical rules, false positives observed in virtual screening can be effectively reduced. There are nine parameters in the input file to set such chemical rules:
A general-purposed set of these chemical rules could be: molecular weight between 200 to 600; LogP between 1 and 5; Number of donor atoms below 6 and number of acceptor atoms below 6. Here logP values are calculated by using the XLOGP2 algorithm.
5. How to interpret the outputs?
All the results will be summarized in a text table. The OUTPUT_MER_FILE parameter in the input file specifies this table. The first line of this table is a title line. Every following line denotes for a single ligand molecule (click here to view an example). The meaning of each column is:
This table is organized in the SYBYL MERGE format. If you open a spreadsheet in SYBYL, you can import this table directly. But since this table is a standard comma-seperated text file, you can also use any other spreadsheet programs, such as Excel and Origin, to load this table.
X-Score also allows you to extract the best-ranked candidates and save each of them in a separate Mol2 file for the convenience of further analysis. The last two parameters in the input file are denoted for this:
There is one more parameter in the input file: CALCULATE_ATOM_BIND_SCORE. It can be set to "YES" or "NO". If it is set to "YES", the program will calculate the contribution of each individual atom to the overall binding affinity of the ligand molecule. These values will be written in the Mol2 file as the atomic charges when the ligand is saved. Therefore, you can inspect them by displaying atomic charges when you view the molecule. Turning on CALCULATE_ATOM_BIND_SCORE has very little impact on the speed of computation.
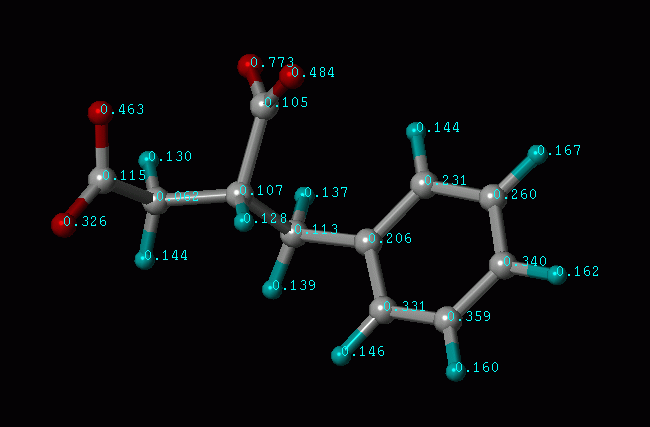
Illustration of Atomic Binding Score (in pKd units)
This "atomic binding score" usually gives you a good idea of which portion of the ligand molecule contributes more to the binding affinity. Accordingly, you can optimize the molecule by enhancing the "good" parts or eliminating the "bad" parts. We, as well as many users, have found this concept useful for structure-based drug design.
6. The shortcut to run X-Score
The standard way for running X-Score, which has been described above, is suitable for scoring multiple ligand molecules against a given target. This is typically seen in a virtual database screening application. But sometimes the user just wants to score one particular ligand against its target and get a fast feedback. X-Score provides a shortcut for this purpose:
xscore the_protein_PDB_file the_ligand_Mol2_file
If a cofactor exists:
xscore the_protein_PDB_file the_cofactor_Mol2_file the_ligand_Mol2_file
In such cases, the following parameters are automatically set by the program as:
The results will be printed on the screen. A Mol2 file which carries the atomic bind scores (saved as atomic charges) is created under your working directory as "xscore.mol2" for further analysis.
That is it! As you can see here, actually X-Score is quite easy to use.
[Content] [Introduction] [Download] [Installation] [Usage] [Trouble Shooting]